Diastereoselektive Synthese von Nukleotid-Prodrugs

Nukleotid-Prodrugs sind Systeme, die es ermöglichen, das Nukleosid Monophosphat in Zellen einzubringen. Aufgrund der hohen Polarität der Nukleotide bei physiologischem pH-Wert sind diese Verbindungen nicht in der Lage, die hydrophobe/lipophile Zellmembran zu durchdringen. Es wurde eine Reihe von Prodrug-Systemen entwickelt, um die beiden negativen Ladungen der Phosphatgruppe zu maskieren. Im Inneren der Zelle sollte die lipophile Maske auf chemischem oder enzymatischem Wege gespalten werden. Das von uns entwickelte CycloSal-Prodrug-System verwendet Salicylalkoholderivate als lipophile Maske (Abbildung 1).[1], [2].
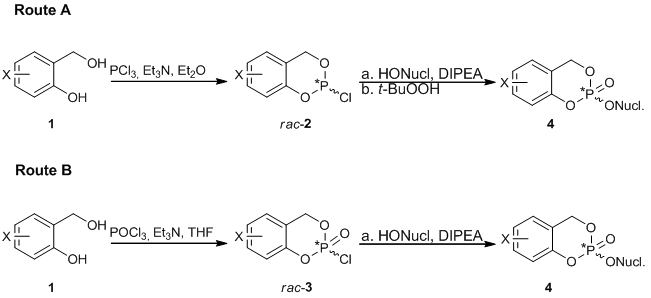 Abbildung 1: Synthese von CycloSal-Pronucleotid 4
Abbildung 1: Synthese von CycloSal-Pronucleotid 4Aufgrund der nicht-stereoselektiven Synthesewege A oder B (Abbildung 1) werden die verfügbaren CycloSal-Prodrugs wegen des neu gebildeten chiralen Zentrums am Phosphoratom immer als Mischungen aus zwei Diastereomeren erhalten. Die beiden Diastereomere haben unterschiedliche chemische und physikalische Eigenschaften und besitzen eine unterschiedliche biologische Aktivität. In einem speziellen Fall wurde festgestellt, dass das RP-Isomer von 3-Methyl-cyclo-Sal-d4TMP eine 10-mal höhere Aktivität als das SP-Isomer aufweist[3].
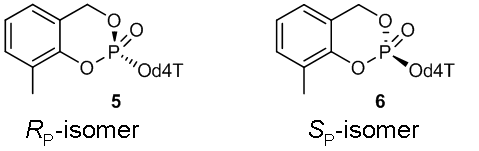 Abbildung 2: (RP)-3-Methyl-cycloSal-d4TMP 5 und (SP)-3-Methyl-cycloSal-d4TMP 6
Abbildung 2: (RP)-3-Methyl-cycloSal-d4TMP 5 und (SP)-3-Methyl-cycloSal-d4TMP 6 Manchmal können die Diastereomere chromatographisch getrennt werden. Dieser Ansatz scheitert jedoch oft. Aus diesem Grund entwickeln wir einen diastereoselektiven Syntheseweg, um diastereomerenreine CycloSal-Prodrugs zu erhalten. Ein synthetischer Ansatz basiert auf der Verwendung des chiralen Hilfsstoffs 7, der eine asymmetrische Induktion am Phosphoratom bewirkt, die zur bevorzugten Bildung des Diastereomers (RP)-8 führt. Die folgende Umwandlung des Diastereomers ergibt die diastereomerenreine CycloSal-Prodrug (SP)-9 (Abbildung 3)[4].
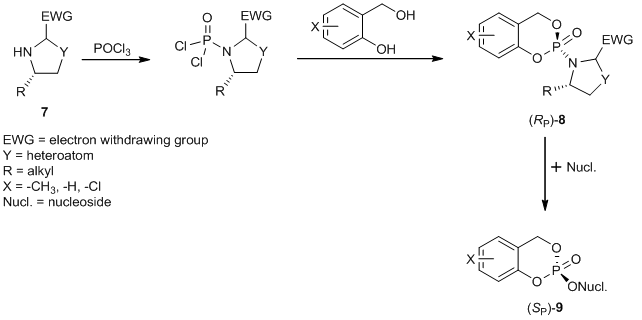 Abbildung 3: Diastereoselektive Synthese von (SP)-cycloSal-Nukleotid-Prodrugs
Abbildung 3: Diastereoselektive Synthese von (SP)-cycloSal-Nukleotid-Prodrugs
Ein anderer synthetischer Ansatz ermöglicht die Synthese nicht nur der (SP)-, sondern auch der (RP)-CycloSal-Prodroge. Das Chloridat rac-3 reagiert mit dem chiralen Hilfsstoff unter Bildung der Diastereomere (SP)-10 und (RP)-8. Nach der Säulenchromatographie der Diastereomere führt jede einzelne Reaktion mit dem Nukleosidanalogon zur diastereomerenreinen CycloSal-Prodrug-Verbindung (Abbildung 4)[5].
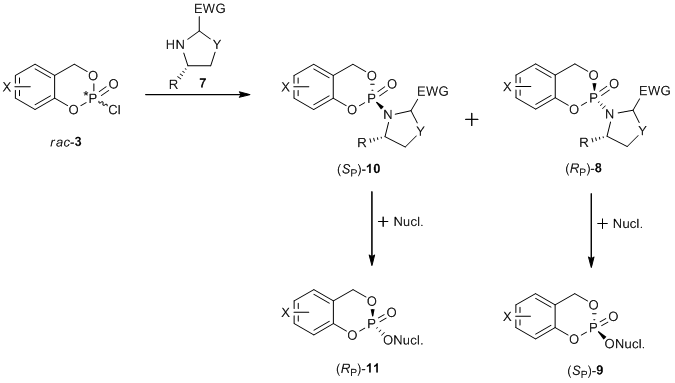 Abbildung 4: Diastereomerspezifische Synthese von diastereomerenreinen CycloSal-Prodrugs.
Abbildung 4: Diastereomerspezifische Synthese von diastereomerenreinen CycloSal-Prodrugs.
Ausgewählte Publikationen
- C. Meier, cycloSal Phosphates as Chemical Trojan Horses for Intracellular Nucleotide and Glycosylmonophosphate Delivery-Chemistry Meets Biology, Eur. J. Org. Chem. 2006, 5, 1081-1102.
- C. Meier, M. Ruppel, D. Vukadinovic, J. Balzarini, „Lock-in“-cycloSal-Pronucleotides- A New Generation of Chemical Trojan Horses?, Mini Rev. Med. Chem. 2004, 4, 383-394.
- C. Meier, cycloSal-Pronucleotides-Design of the Concept, Chemistry, and Antiviral Activity, Advances in Antiviral Drug Design 2004, 4, 147-213.
- a) E. H. Rios Morales, C. Arbelo Román, J. O. Thomann, C. Meier, Linear Synthesis of Chiral cycloSal-Pronucleotides, Eur. J. Org. Chem. 2011, 4397-4408. b) E. H. Rios Morales, J. Balzarini, C. Meier, Diastereoselective Synthesis of cycloSaligenyl-Nucleosyl-Phosphotriesters, Chem. Eur. J. 2011, 17, 1649-1659
- E. H. Rios Morales, J. Balzarini, C. Meier, Stereoselective Synthesis and Antiviral Activity of Methyl-Substituted cycloSal-Pronucleotides, J. Med. Chem. 2012, 55, 7245-7252.