Design von lipophilen trojanischen Pferden für die intrazelluläre Abgabe von phosphorylierten Nukleosidmetaboliten

1. The cycloSal-technology
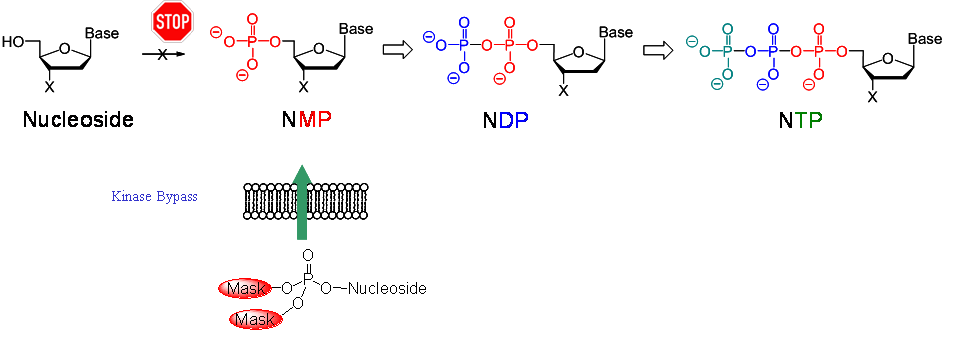
Nukleosidanaloga sind wichtige Verbindungen zur Bekämpfung von Virusinfektionen. Sie sind als solche jedoch nicht bioaktiv, sondern erfordern eine intrazelluläre Aktivierung durch Wirtszellkinasen. Diese Enzyme wandeln das Nukleosid über das Mono- und das Diphosphat in das letztlich aktive Nukleosidtriphosphat um. Da es sich hierbei nur um ein Analogon der natürlichen Nukleoside handelt, ist der Metabolismus oft ineffizient und manchmal findet die Phosphorylierung überhaupt nicht statt. Im letzteren Fall ist das Nukleosidanalog völlig inaktiv. Ein Versuch, die Bioaktivierung zu verbessern, könnte jedoch der Entwurf von lipophilen Vorläufern der phosphorylierten Metabolite sein, die in die Zellen gelangen und die phosphorylierten Metabolite nach einem bestimmten Mechanismus entweder auf chemischem oder enzymatischem Wege liefern können.
In diesem Zusammenhang haben wir den so genannten CycloSal-Pronukleotid-Ansatz entwickelt, der zu den weltweit führenden Vornukleotid-Systemen gehört.[1]
Die Grundidee ist ein hochselektiver, chemisch getriebener Hydrolysemechanismus, der Nukleotide in Zellen liefert. Als lipophile Maskierungseinheit wird ein Salicylalkohol kovalent an die beiden verbleibenden Säurefunktionalitäten des Nukleotids gebunden. Durch den intelligenten Aufbau dieses Systems hinterlässt der erste Hydrolyseschritt ein hochreaktives Zwischenprodukt, das sich spontan aufspaltet und das Nukleotid liefert (Verbindungen der ersten Generation). Der Abgabemechanismus führt selektiv zu Nukleotiden, die die bekannten pseudorotationalen Prozesse am Phosphoratom vermeiden. Der im folgenden Schema dargestellte Mechanismus ist durch 31P-NMR-Studien, HPLC-basierte Studien sowie Isotopenmarkierung nachgewiesen und wurde auch in kinetischen Studien charakterisiert.[2-4]
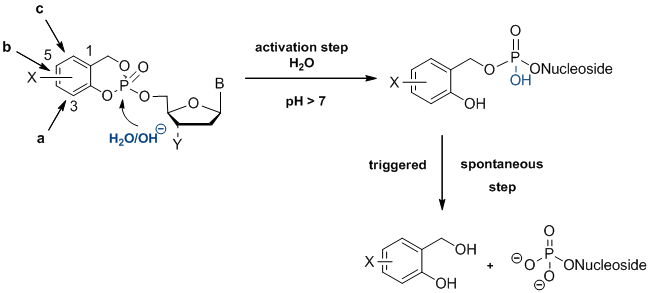 Abbildung 2: Hydrolysemechanismus von CycloSal-Pronukleotiden.
Abbildung 2: Hydrolysemechanismus von CycloSal-Pronukleotiden.
Der Ansatz wurde erfolgreich auf verschiedene Nukleosidanaloga angewandt, die beweisen, dass die antivirale Aktivität verbessert, die Resistenz gegen ein parentales Nukleosid überwunden und sogar inaktive Nukleosidanaloga in bioaktive Derivate umgewandelt werden können.[5-9]
Aufgrund des lipophilen Charakters der CycloSal-Phosphat-Triester und ihres chemisch ausgelösten Abgabemechanismus wird ein Gleichgewicht der Wirkstoffkonzentration durch die Zellmembran gebildet. Um die lipophilen CycloSal-Triester in den Zellen einzufangen, wurden so genannte "Lock-in"-CycloSal-Pronukleotide entwickelt (Verbindungen der zweiten Generation). Diese Verbindungen tragen Esterase-abspaltbare Ester, die an den aromatischen Ring gebunden sind. Um jeglichen Einfluss auf die chemischen Hydrolyseeigenschaften zu vermeiden, werden die Esteranteile durch einen C2-Spacer vom aromatischen Ring getrennt.. CycloSal-d4TMP-Säureester und CycloSal-d4TMP-Alkoholester[10] sowie acyl[11] oder aminosäureester[12] funktionalisierte CycloSal-d4TMPs wurden untersucht. Hydrolysestudien in Phosphatpuffer (PBS, pH=7,3) und in T-Lymphozyten-CEM-Zellextrakten ergaben, dass ein intrazelluläres Trapping möglich ist, wenn hochpolare, geladene CycloSal-d4TMP-Säuren freigesetzt werden. Die schnelle intrazelluläre Freisetzung dieser Verbindungen ist das primäre Ziel des "Lock-in"-Konzepts. Als nächstes wird D4TMP durch chemische Hydrolyse aus den CycloSal-d4TMP-Säuren freigesetzt. Es wurde jedoch festgestellt, dass diese Freisetzung sehr langsam erfolgt.
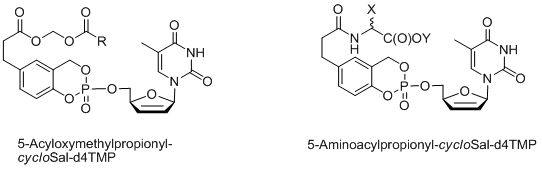
Deshalb wurden als konzeptionell andere Extension enzymatisch aktivierte CycloSal-Pronukleotide entwickelt (Prodrugs der dritten Generation). Bei diesem Ansatz haben Prodrugs ebenfalls einen lipophilen, funktionalisierten Substituenten mit elektronenspendenden oder nur schwach elektronenziehenden Eigenschaften an den aromatischen Ring der cycloSal-Maske gebunden. Nach passivem Transport in Zellen wird der funktionalisierte Substituent durch enzymatische Spaltung in eine starke elektronenziehende Gruppe umgewandelt. Diese Gruppe bewirkt eine signifikante Abnahme der Hydrolysestabilität der cycloSal-Verbindung. Im Gegensatz zum "Lock-in"-Ansatz führt die enzymatische Auslösung in diesem Fall zu einer schnellen Abgabe des Nukleotids, statt zu einem hochpolaren, chemisch stabileren Zwischenprodukt. Damit wird die Fallenidee bei diesen Verbindungen auf der Nukleotidebene realisiert.
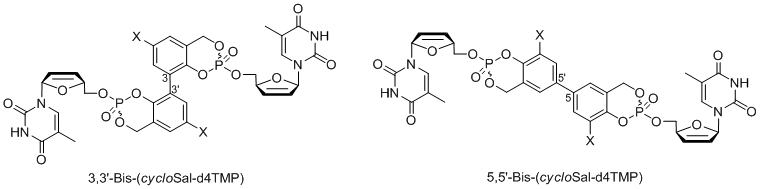
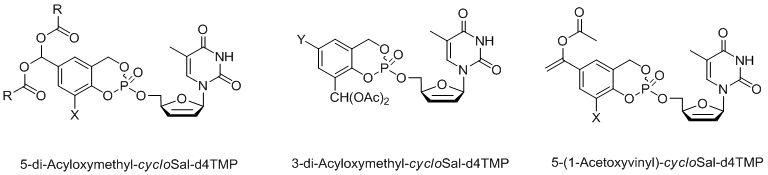 Abbildung 4: Allgemeine Struktur von enzymatisch aktivierten CycloSal-Pronukleotiden.
Abbildung 4: Allgemeine Struktur von enzymatisch aktivierten CycloSal-Pronukleotiden.Als enzymatisch aktivierte CycloSal-Prodrugs wurden sowohl verschiedene Diacyloxymethyl-cycloSal-d4TMPs[13] als auch 5-(1-Acetoxyvinyl)-cycloSal-d4TMPs[14] entwickelt. Kürzlich haben wir den CycloSal-Pronukleotid-Ansatz auf die erste Synthese so genannter "hochbelasteter" Pronukleotide (Verbindungen der vierten Generation) ausgedehnt[15,16]. Im ursprünglichen Konzept betrug das Verhältnis der Maskierungseinheit zum Nukleotid 1:1. Um dieses Verhältnis zu erhöhen, so dass mehr Nukleotide an den Vektor gebunden sind, wurden Tetrole oder Dimere von Salicylalkoholen verwendet und mit zwei Äquivalenten des entsprechenden Nukleosids geladen. Es konnte gezeigt werden, dass solche Verbindungen durch chemische Hydrolyse tatsächlich stufenweise zwei Nukleotidmoleküle liefern. Diese Verbindungen erwiesen sich auch in einer mutierten T-Lymphozyten-Zelllinie, die für die Anti-HIV-Tests verwendet wird, als aktiv.
Abbildung 5: Allgemeine Struktur von "hochbelasteten"-CycloSal-Pronukleotiden.
2. Prodrugs von Nukleosiddiphosphaten
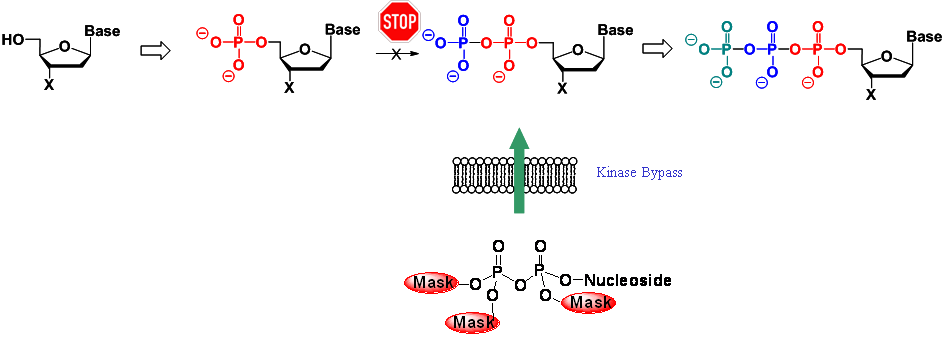 Abbildung 6: Allgemeiner Metabolismus von Nukleosidanaloga und Kinase-Umgehung von Nukleosiddiphosphaten.
Abbildung 6: Allgemeiner Metabolismus von Nukleosidanaloga und Kinase-Umgehung von Nukleosiddiphosphaten.
Im Gegensatz zu Prodrugs von Nukleotiden ist die Entwicklung von Nukleosiddiphosphat- oder sogar Triphosphat-Prodrugs nur in sehr wenigen Fällen angegangen worden. Dies ist bemerkenswert, denn es ist bekannt, dass z.B. 3á-Azido-3á-deoxythymidin (AZT), das erste zugelassene nukleosidische Anti-HIV-Medikament, nur sehr langsam durch Thymidylatkinase diphosphoryliert wird. Die daraus resultierende Anhäufung von AZT-Monophosphat (AZTMP) ruft zudem schwere Nebenwirkungen hervor.
Kürzlich haben wir über den ersten erfolgreichen Nukleosiddiphosphat-Prodrug-Ansatz berichtet.[17] Im Gegensatz zur CycloSal-Technologie beruht hier der Abgabemechanismus auf einem enzymatisch ausgelösten Prozess. Chemische Methoden führten nicht zu einer selektiven Freisetzung des entsprechenden Nukleosiddiphosphats. Bis-(acyloxybenzyl)ester (BAB-Ester) des Nukleosiddiphosphats wurden hergestellt und zeigten sehr vielversprechende und aufregende Ergebnisse bezüglich der bioreversiblen Deprotektion dieser Klasse hochpolarer Metabolite. Die Leitstruktur dieser Prodrugs sowie der vorgeschlagene Hydrolysemechanismus sind im folgenden Schema dargestellt.
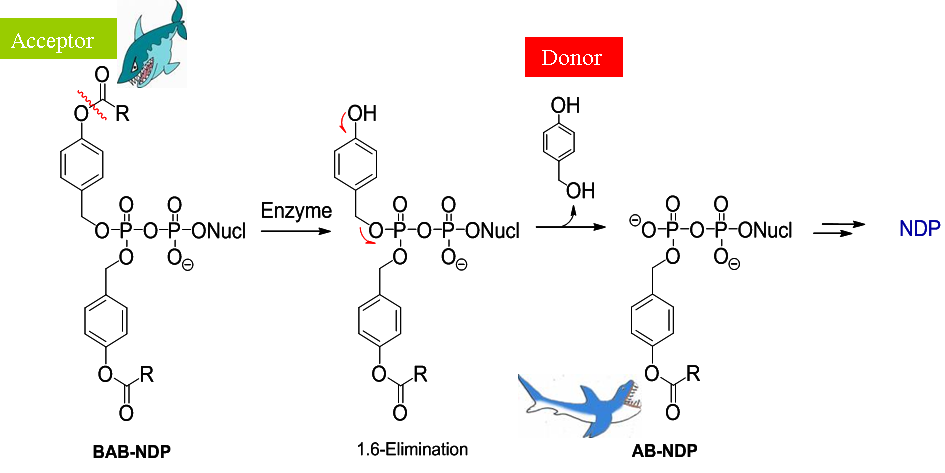 Abbildung 7: Enzymatische Spaltung von Bis-BAB-Nukleosiddiphosphat-Prodrugs.
Abbildung 7: Enzymatische Spaltung von Bis-BAB-Nukleosiddiphosphat-Prodrugs.Wir arbeiten derzeit an der Optimierung dieses anspruchsvollen und vielversprechenden Konzepts im Hinblick auf Hydrolyseeigenschaften, Lipophilie verschiedener Zielstrukturen und Anwendung des Konzepts auf verschiedene biologisch aktive Nukleoside.
Artikel in der Zeitung "Die Welt" über Nukleosiddiphosphat-Prodrugs (03.11.2008):
http://www.welt.de/welt_print/article2664791/Trojanische-Pferde-gegen-Aids-und-Hepatitis.html
Ausgewählte Publikationen:
- C. Meier, CycloSal-Phosphates as Chemical Trojan Horses for the Intracellular Nucleotide and Glycosylmonophosphate Delivery – Chemistry meets Biology, Eur. J. Org. Chem. 2006, 1081-1102.
- C. Meier, A. Meerbach, J. Balzarini, cycloSal-Pronucleotides – Development of First and Second Generation Chemical Trojan Horses for Antiviral Chemotherapy, Frontiers in Bioscience 2004, 9, 873-890.
- C. Meier, M. Ruppel, D. Vukadinovic, J. Balzarini, “Lock-in”-cycloSal-pronucleotides – A New Generation of Chemical Trojan Horses, Minirev. Med. Chem. 2004, 4, 383-394.
- J. Balzarini, S. Aquaro, T. Knispel, C. Rampazzo, V. Bianchi, C.-F. Perno, E. De Clercq, C. Meier, CycloSaligenyl-2‘,3‘-Didehydro-2‘,3‘-dideoxythymidine Monophosphate (cycloSal-d4TMP): Efficient Intracellular Delivery of d4TMP, Mol. Pharmacol. 2000, 58, 928-935.
- C. Meier, A. Lomp, A. Meerbach, P. Wutzler, CycloSal-BVDUMP-Pronucleotides – How to Convert an anti-EBV-inactive Nucleoside Analogue into a Bioactive Compound, J. Med. Chem. 2002, 45, 5157-5172.
- C. Meier, T. Knispel, E. De Clercq, J. Balzarini, CycloSal-Pro-Nucleotides (cycloSal-NMP) of 2',3'-dideoxyadenosine (ddA) and 2',3'-dideoxy-2',3'-didehydroadenosine (d4A): Synthesis and Antiviral Evaluation of a Highly Efficient Nucleotide Delivery System, J. Med. Chem. 1999, 42, 1604-1614.
- C. Meier, T. Knispel, V. E. Marquez, M. A. Siddiqui, E. De Clercq, J. Balzarini, CycloSal-Pro-Nucleotides of 2'-fluoro-ara- and 2'-fluoro-ribo-2',3'-dideoxyadenosine (F-ara- and F-ribo-ddA) as a Strategy to Bypass a Metabolic Blockade, J. Med. Chem. 1999, 42, 1615-1624.
- A. Sauerbrei, C. Meier, A. Meerbach, P. Wutzler, Inhibition Efficiency of cycloSal-Nucleoside Monophophates of Aciclovir and Brivudine on DNA Synthesis of Orthopoxviruses, Antiviral Chem. Chemother. 2006, 17, 25-31.
- C. Meier, U. Görbig, C. Müller, J. Balzarini, CycloSal-PMEA and cycloAmb-PMEA – Potentially new Phosphonate Prodrugs on the Basis of the cycloSal-Pronucleotide Approach, J. Med. Chem. 2005, 48, 8079-8086.
- C. Meier, M. F. H. Ruppel, D. Vukadinović, J. Balzarini, Second Generation of cycloSal-Pronucleotides with Esterase-Cleavable Sites: The „Lock-in“-Concept, Nucleosides, Nucleotides Nucleic Acids 2004, 23, 89-115.
- C. Meier, C. Ducho, H. Jessen, D. Vukadinović-Tenter, J. Balzarini, Second-Generation cycloSal-d4TMP Pronucleotides Bearing Esterase-Cleavable Sites – The „Trapping“ Concept, Eur. J. Org. Chem. 2006, 197-206.
- H. J. Jessen, J. Balzarini, C. Meier, Intracellular Trapping of cycloSal-Pronucleotides: Modification of Prodrugs with Amino Acid Esters, J. Med. Chem. 2008, 51, 6592-6598.
- N. Gisch, J. Balzarini, C. Meier, Studies on Enzyme-cleavable Dialkoxymethyl-cycloSaligenyl-2’,3’-dideoxy-2’,3’-didehydrothymidine Monophosphates, J. Med. Chem. 2008, 51, 6752-6760.
- N. Gisch, F. Pertenbreiter, J. Balzarini, C. Meier, 5-(1-Acetoxyvinyl)-cycloSaligenyl-2’,3’-dideoxy-2’,3’-didehydrothymidine Monophosphates – A Second Type of New, Enzymatically Activated cycloSal-Pronucleotides, J. Med. Chem. 2008, 51, 8115-8123.
- C. Ducho, U. Görbig, S. Jessel, N. Gisch, J. Balzarini, C. Meier, Bis-cycloSal-d4T-monophosphates – Drugs that Deliver Two Molecules of Bioactive Nucleotides, J. Med. Chem. 2007, 50, 1335-1346.
- N. Gisch, J. Balzarini, C. Meier, Doubly loaded cycloSaligenyl-Pronucleotides – 5,5’-Bis-(cycloSaligenyl-2’,3’-dideoxy-2’,3’-didehydrothymidine monophosphates), J. Med. Chem. 2009, 52, 3464-3473.
- H. J. Jessen, T. Schulz, J. Balzarini, C. Meier, Bioreversible protection of nucleosidediphosphates, Angew. Chem. Int. Ed. Engl. 2008, 47, 8719-8722.