Stereoselektive Synthesen potenziell antiviral wirksamer carbozyklischer Nukleoside

Was sind carbocyclische Nukleoside?
Carbocyclische Nukleoside sind Nukleosidanaloga, bei denen die Ribose-Einheit durch einen Cyclopentanring ersetzt ist.
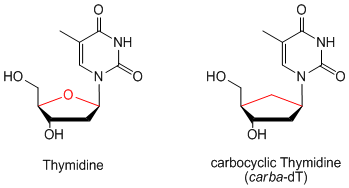
Dies führt zu einer erhöhten Hydrolysestabilität gegenüber Pyrophosphorylasen, die normalerweise glykosidische Bindungen spalten. Der Ersatz des aminalen Sauerstoffs durch eine Methylengruppe wirkt sich auch auf die Konformation des fünfgliedrigen Rings aus. Aufgrund ihrer ungewöhnlichen Ringfaltenbildung haben carbocyclische Nukleoside im Vergleich zu natürlichen Nukleosiden eine andere Struktur-Wirkungs-Beziehung (SAR)[1] und können daher auch potenziell neue biologische Eigenschaften aufweisen:[2,3]
- verbesserte enzymatische Resistenz
verbesserte Hydrolysestabilität
reduzierte Zytotoxizität
Was sind unsere ZIele?
Neben der Entwicklung effizienter, flexibler und stereoselektiver Synthesestrategien für die Herstellung von carbocyclischen Nukleosiden ist es das Ziel unserer Arbeit, neue potentiell biologisch aktive carbocyclische Nukleoside zu finden und carbocyclische Derivate etablierter antiviral wirksamer Medikamente zu synthetisieren. Zum Beispiel wurde festgestellt, dass carbocyclisches Thymidin (D-Carba-dT) ein effizienter Inhibitor viraler Vektoren ist, die NRTI-resistente HIV-1 reverse Transkriptase replizieren. Interessanterweise folgt die Hemmung einem neuen Mechanismus, der zu einem so genannten verzögerten Abbruch der DNA-Kette führt.[4]
Wie erhält man carbocyclische Nukleoside?
Carbocyclische Nukleoside sind synthetisch die anspruchsvollste Klasse von Nukleosiden, die mehrstufige und oft aufwendige Synthesewege erfordern, um die notwendige Stereochemie einzuführen. Es gibt zwei Hauptstrategien für die Herstellung von carbocyclischen Nukleosiden. Beim linearen Ansatz wird ein Cyclopentylamin als Ausgangsmaterial verwendet, und der Heterozyklus wird schrittweise aufgebaut (siehe Schema 1).
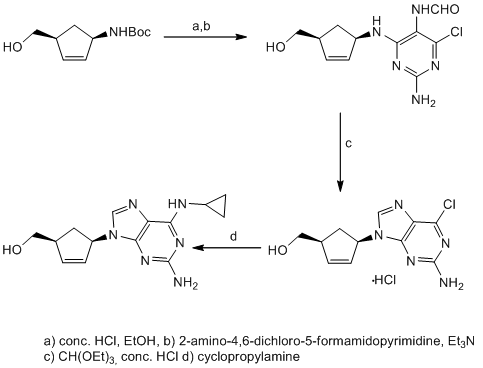
Die flexiblere Strategie ist ein konvergenter Ansatz: eine funktionalisierte carbocyclische Einheit wird mit einem Heterocyclus kondensiert, was schnell zu einer Vielzahl von carbocyclischen Nukleosiden führt. Ursprünglich begannen wir unsere Synthesen aus Cyclopentadien 1, das deprotoniert und mit Benzyloxymethylchlorid alkyliert wird, um das Dien 2 zu erhalten. Dieses Material wird durch eine Hydroborierung in Cyclopentenol 3 umgewandelt oder in zwei thermodynamisch stabilere Cyclopentadiene 4a,b isomerisiert. Mit dem Schutz und einem weiteren Hydroborierungsschritt zu 5 erhalten wir Zugang zu einer enantiomerenreinen Vorstufe für die Synthese einer Vielzahl von carbocyclischen 2'-Desoxynukleosiden, z.B.: carba-dT, carba-dA oder carba-BVDU.[6] Die isomeren Diene 4a,b wurden an den racemischen carbocyclischen Rest 6 hydroboriert.
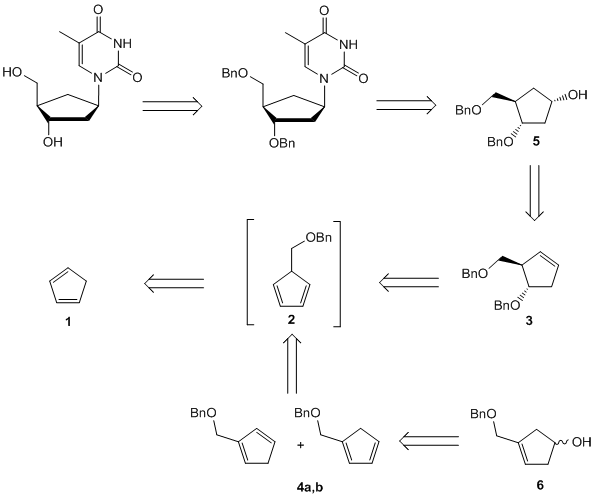 Schema 2: Konvergenter Ansatz für die Synthese von Carba-dT.
Schema 2: Konvergenter Ansatz für die Synthese von Carba-dT.Der asymmetrische Syntheseweg und der oben beschriebene racemische Weg sind kurze und effiziente Wege zu diversen carbocyclischen D- oder L-Nukleosiden (Schema 2). Verschiedene Heterocyclen können zu diesen Vorläufern kondensiert werden, was zu carbocyclischen Purin- und Pyrimidin-Nukleosiden führt. Neben α- und β-Nukleosiden waren auch carbocyclische Epi- und Iso-Nukleoside in der 2'-Desoxyxylose-Form zugänglich.[7]
Was ist noch möglich? Das racemische Cyclopentenol 6 kann durch eine modifizierte Mitsunobu-Reaktion gekoppelt werden. Darüber hinaus bietet diese Strategie die Möglichkeit, neue carbocyclische Nukleoside zu synthetisieren, indem die Doppelbindung vor oder nach der Einführung der Nukleobase funktionalisiert wird (Schema 3).[8]
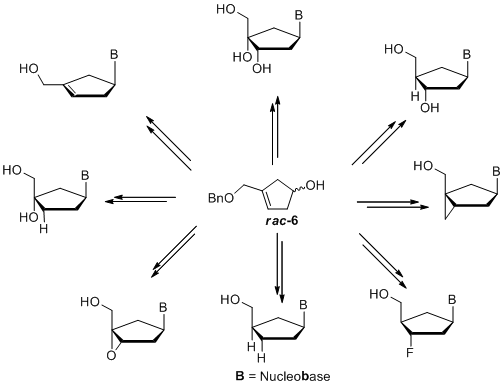
Andere interessante carbocyclische Vorläufer wie Cyclopentenol 7 können zur Synthese mehrerer Klassen von carbocyclischen Nukleosidanaloga verwendet werden, z.B.: 2',3'-Dideoxy-2',3'-didehydro-Nukleoside (d4-Nukleoside), 2',3'-Dideoxynukleoside (ddNs), Ribonukleoside, bicyclische Nukleoside oder sogar 2'-Fluornukleoside.
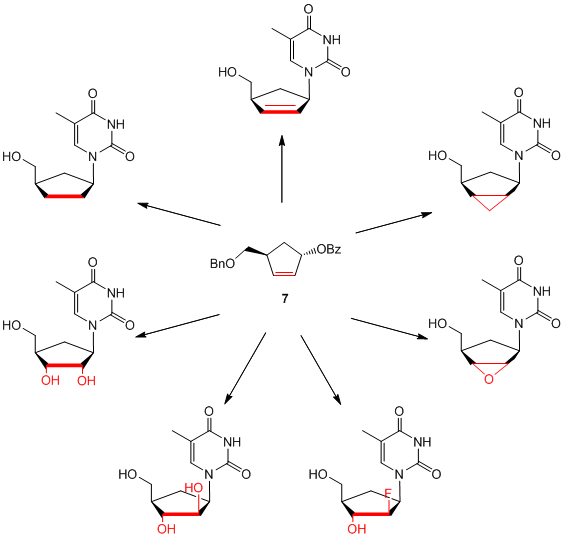 Schema 4: Funktionalisierte carbocyclische Thymidinanaloga auf der Basis von Cyclopentenol 7.
Schema 4: Funktionalisierte carbocyclische Thymidinanaloga auf der Basis von Cyclopentenol 7.
[1] V. E. Marquez, T. Ben-Kasus, J. J. Barchi, K. M. Green, M .C. Nicklaus, R. Agbaria, J. Am. Chem. Soc. 2004,126, 543.
[2] A. D. Borthwick, K. Biggadike, Tetrahedron 1992, 48, 571.
[3] H. Bricaud, P. Herdewijn, E. De Clercq, Biochem. Pharmacol. 1983, 3583.
[4] P. L. Boyer, B. C. Vu, Z. Ambrose, J. G. Julias, S. Warnecke, C. Liao, C. Meier, V. E. Marquez, S. H. Hughes, J. Med. Chem. 2009, 52, 5356.
[5] S. M. Daluge, M. T. Martin, B. R. Sickles, D. A. Livingston, Nucleosides, Nucleotides Nucleic Acids 2000, 19, 297.
[6] O. R. Ludek, C. Meier, Synthesis 2003, 2101.
[7] O. R. Ludek, T. Kraemer, J. Balzarini, C. Meier, Synthesis 2006, 1313.
[8] M. Mahler, B. Reichardt, P. Hartjen, J. van Lunzen, C. Meier, Chem. Eur. J. 2012, 18, 11046-11062.