Stereoselective Syntheses of Potentially Antivirally Active Carbocyclic Nucleosides

What are carbocyclic nucleosides?
Carbocyclic nucleosides are nucleoside analogues, in which the ribose moiety is replaced by a cyclopentane ring.
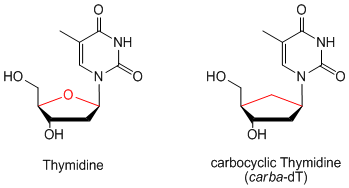
This leads to enhanced hydrolysis stability towards pyrophosphorylases, which normally cleave glycosidic bonds. The replacement of the aminal oxygen by a methylene group also effects the conformation of the five-membered ring. Due to their unusual ring-puckering carbocyclic nucleosides have a different structure-activity-relationship (SAR) compared to natural nucleosides.[1] Therefore, they may also exhibit potentially new biological properties:[2,3]
- improved enzymatic resistance
- enhanced hydrolysis stability
- reduced cytotoxicity
What are our objectives?
Beside developing efficient, flexible and stereoselective synthetic strategies for the preparation of carbocyclic nucleosides, the aim of our work is to find new potentially biologically active carbocyclic nucleosides and to synthesize carbocyclic derivatives of established antivirally active drugs. For example carbocyclic thymidine (D-carba-dT) was found to be an efficient inhibitor of viral vectors that replicate NRTI-resistant HIV-1 reverse transcriptase. Interestingly, the inhibition follows a new mechanism, which results in a so called delayed DNA-chain termination.[4]
How to obtain carbocyclic nucleosides?
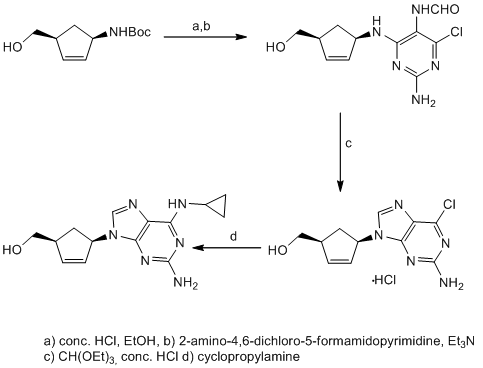
Carbocyclic nucleosides are synthetically the most challenging class of nucleosides, requiring multi-step and often elaborate synthetic pathways to introduce the necessary stereochemistry. There are two main strategies for the preparation of carbocyclic nucleosides. In the linear approach a cyclopentylamine is used as starting material and the heterocycle is built in a stepwise manner (see Scheme 1).
The more flexible strategy is a convergent approach: a functionalized carbocyclic moiety is condensed with a heterocycle rapidly leading to a variety of carbocyclic nucleosides. Initially, we started our syntheses from cyclopentadiene 1 that is deprotonated and alkylated with benzyloxymethyl chloride to give the diene 2. This material is converted by a hydroboration into cyclopentenol 3 or isomerized into two thermodynamically more stable cyclopentadienes 4a,b. With the protection and another hydroboration step to 5 we gain access to an enantiomerically pure precursor for the synthesis of a variety of carbocyclic 2’-deoxynucleosides e.g.: carba-dT, carba-dA or carba-BVDU.[6] The isomeric dienes 4a,b were hydroborated to the racemic carbocyclic moiety 6.
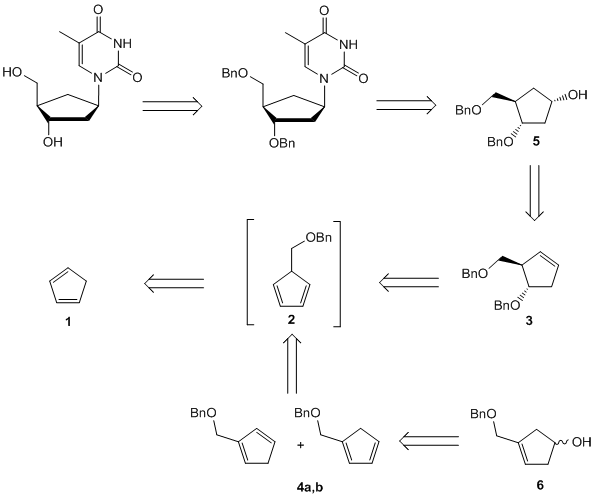
The asymmetric synthesis route and the racemic route above are short and efficient ways to diverse carbocyclic D- or L-nucleosides (Scheme 2). Different heterocycles can be condensed to these precursors leading to carbocyclic purine- and pyrimidine-nucleosides. Beside α- and β-nucleosides, carbocyclic epi- and iso-nucleosides in the 2’-deoxyxylose form were accessable.[7]
What else is possible? The racemic cyclopentenol 6 can be coupled by a modified Mitsunobu-reaction. Moreover, this strategy offers the possibility of synthesizing new carbocyclic nucleosides by functionalizing the double bond before or after introduction of the nucleobase (scheme 3).[8]
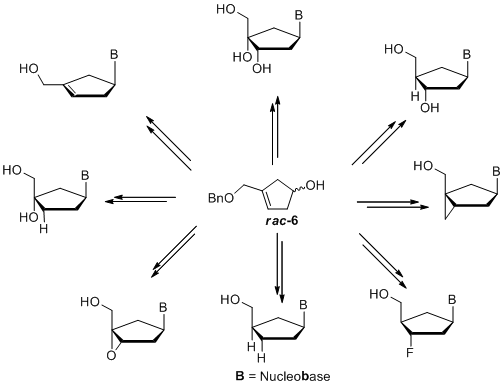
Other interesting carbocyclic precursors like cyclopentenol 7 can be used to synthesize several classes of carbocyclic nucleoside analogues, e.g.: 2’,3’-dideoxy-2’,3’-didehydro nucleosides (d4-nucleosides), 2’,3’-dideoxynucleosides (ddNs), ribonucleosides, bicyclic nucleosides or even 2’-fluoro-nucleosides.
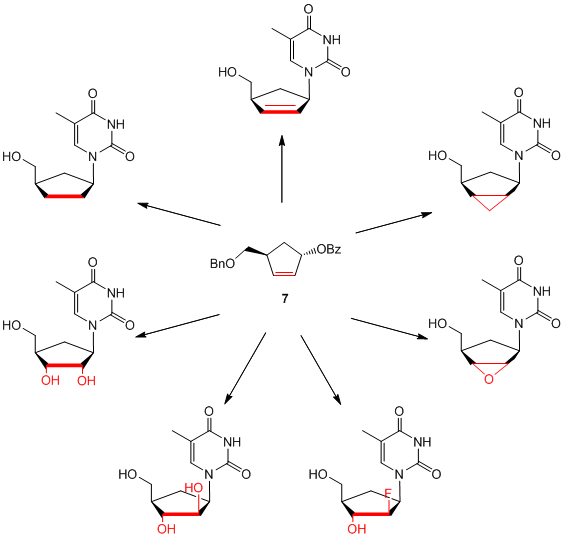 Scheme 4: Functionalized carbocyclic thymidine analogues based on cyclopentenol 7.
Scheme 4: Functionalized carbocyclic thymidine analogues based on cyclopentenol 7.
[1] V. E. Marquez, T. Ben-Kasus, J. J. Barchi, K. M. Green, M .C. Nicklaus, R. Agbaria, J. Am. Chem. Soc. 2004,126, 543.
[2] A. D. Borthwick, K. Biggadike, Tetrahedron 1992, 48, 571.
[3] H. Bricaud, P. Herdewijn, E. De Clercq, Biochem. Pharmacol. 1983, 3583.
[4] P. L. Boyer, B. C. Vu, Z. Ambrose, J. G. Julias, S. Warnecke, C. Liao, C. Meier, V. E. Marquez, S. H. Hughes, J. Med. Chem. 2009, 52, 5356.
[5] S. M. Daluge, M. T. Martin, B. R. Sickles, D. A. Livingston, Nucleosides, Nucleotides Nucleic Acids 2000, 19, 297.
[6] O. R. Ludek, C. Meier, Synthesis 2003, 2101.
[7] O. R. Ludek, T. Kraemer, J. Balzarini, C. Meier, Synthesis 2006, 1313.
[8] M. Mahler, B. Reichardt, P. Hartjen, J. van Lunzen, C. Meier, Chem. Eur. J. 2012, 18, 11046-11062.