Design of lipophilic trojan horses for the intracellular delivery of phosphorylated nucleoside metabolites

1. The cycloSal-technology
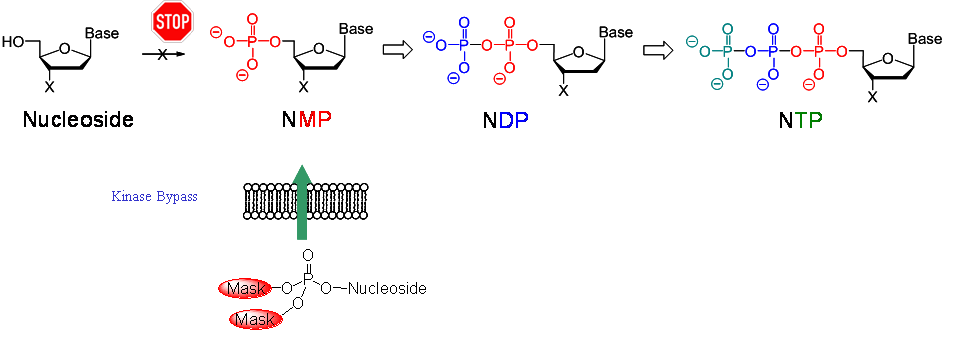
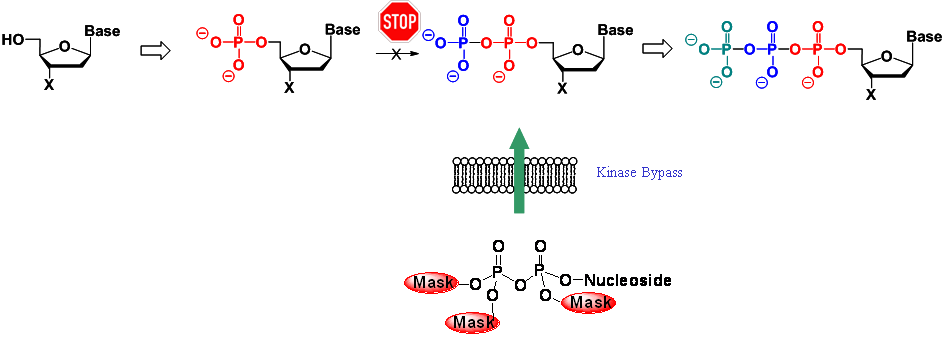
Nucleoside analogues are important compounds to combat viral infections. However, nucleoside analogues are not bioactive as such but they require intracellular activation by host cell kinases. These enzymes convert the nucleoside via the mono- and the diphosphate into the ultimately active nucleoside triphosphate. Due to the characteristic of being only an analogue of the natural nucleosides, the metabolism often is inefficient and sometimes the phosphorylation does not occur at all. In the latter scenario, the nucleoside analogue will be completely inactive. However, an attempt to improve the bioactivation could be the design of lipophilic precursors of the phosphorylated metabolites that are able to enter cells and deliver the phosphorylated metabolite following a specific mechanism either by chemical or by enzymatic means.
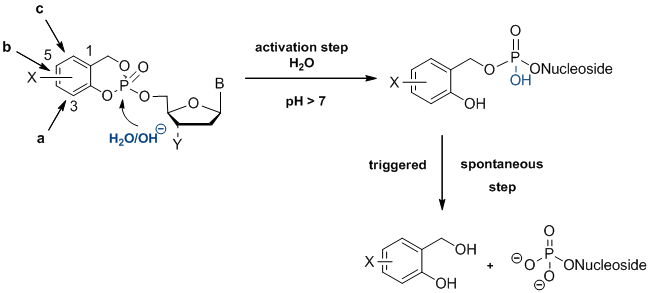
In this context we have developed the so-called cycloSal-pronucleotide approach that is one of the leading pronucleotide systems world-wide.[1] The basic idea is a highly selective chemically driven hydrolysis mechanism that delivers nucleotides into cells. As lipophilic masking unit a salicylalcohol is covalently attached to the two remaining acid functionalities of the nucleotide. By intelligent construction of this system, the first hydrolysis step leaves a highly reactive intermediate that spontaneously breaks down to yield the nucleotide (first generation compounds). The delivery mechanism leads selectively to nucleotides avoiding the known pseudorotational processes at the phosphorus atom. The mechanism depicted in the following scheme has been proven by 31P-NMR studies, HPLC-based studies as well as isotope-labelling and was characterized in kinetic studies as well.[2-4]
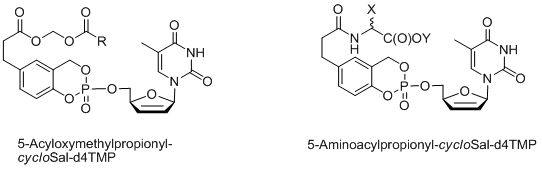
The approach has been applied successfully to various nucleoside analogues proving that the antiviral activity can be improved, resistance towards a parent nucleoside can be overcome and even inactive nucleoside analogues can be converted into bioactive derivatives.[5-9] Due to the lipophilic character of cycloSal-phosphate triesters and their chemically-triggered delivery mechanism, a drug concentration equilibrium is formed through the cell membrane. In order to trap the lipophilic cycloSal-triesters inside cells, so called “lock-in”-cycloSal-pronucleotides were designed (second generation compounds). These compounds are bearing esterase-cleavable esters attached to the aromatic ring. To avoid any influence on the chemical hydrolysis properties, the ester moieties are separated from the aromatic ring by a C2-spacer. CycloSal-d4TMP acid ester and cycloSal-d4TMP alcohol ester[10] as well as acylal[11] or amino acid ester[12] functionalized cycloSal-d4TMPs have been investigated. Hydrolysis studies in phosphate buffer (PBS, pH=7.3) and in T-lymphocyte CEM cell extracts revealed that an intracellular trapping is possible, if highly polar, charged cycloSal-d4TMP acids are released. The fast intracellular release of these compounds is the primary aim of the “lock-in”-concept. D4TMP is released from the cycloSal-d4TMP acids by chemical hydrolysis next. However, this release was found to be very slow.
Therefore, as a conceptually different extension, enzymatically activated cycloSal-pronucleotides have been developed (third generation prodrugs). In this approach, prodrugs also have a lipophilic, functionalized substituent with electron-donating or only weak electron-withdrawing properties attached to the aromatic ring of the cycloSal-mask. After passive transport into cells the functionalized substituent is converted into a strong electron-withdrawing group by enzymatic cleavage. This group causes a significant decrease in hydrolysis stability of the cycloSal-compound. In contrast to the “lock-in” approach, in this case the enzymatic triggering results in a fast delivery of the nucleotide, instead of a highly polar, chemically more stable intermediate. Thus, the trapping idea with these compounds is realized on the nucleotide level.
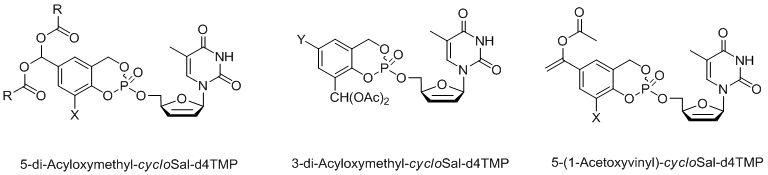
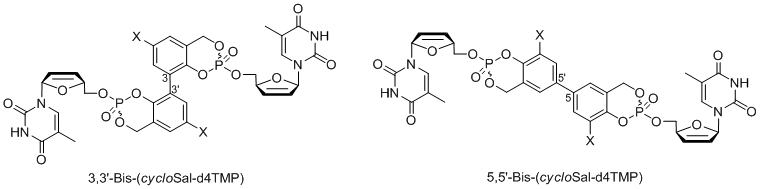
As enzymatically activated cycloSal-prodrugs different diacyloxymethyl-cycloSal-d4TMPs[13] as well as 5-(1-acetoxyvinyl)-cycloSal-d4TMPs[14] have been developed. Recently, we have extended the cycloSal-pronucleotide approach to the first synthesis of so-called “high-loaded” pronucleotides (fourth generation compounds).[15,16] In the original concept the ratio of the masking unit to the nucleotide was 1:1. In order to increase this ratio so that more nucleotides are attached to the vector, tetrols or dimers of salicylalcohols were used and charged with two equivalents of the corresponding nucleoside. It was shown that such compounds indeed deliver step-wise two nucleotide molecules by chemical hydrolysis. These compounds also proved to be active in a mutant T-lymphocyte cell line that is used for the anti-HIV tests.
Figure 5: General structure of "high-loaded"-cycloSal-pronucleotides.
2. Prodrugs of Nucleoside diphosphates
In contrast to prodrugs of nucleotides, the development of nucleoside diphosphate or even triphosphate prodrugs has only been addressed in very few cases. This is remarkable, because it is known that e.g. 3á-azido-3á-deoxythymidine (AZT), the first approved nucleosidic anti-HIV drug, is only very slowly diphosphorylated by thymidylate kinase. Further, the resulting accumulation of AZT monophosphate (AZTMP) provokes severe side-effects.
Recently, we have reported on the first successful nucleoside diphosphate prodrug approach.[17] In contrast to the cycloSal-technology, here the delivery mechanism relies on an enzymatically triggered process. Chemical methods did not lead to a selective release of the corresponding nucleoside diphosphate. Bis-(acyloxybenzyl)esters (BAB-esters) of the nucleoside diphosphate were prepared and showed very promising and exciting results referring to the bioreversible deprotection of this class of highly polar metabolites. The lead structure of these prodrugs is shown in the scheme below as well as the proposed mechanism of hydrolysis.
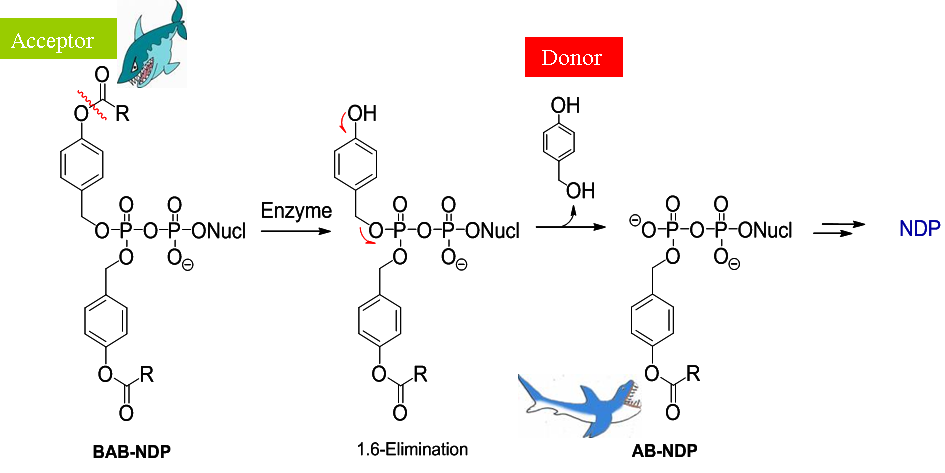
We are currently working on the optimization of this challenging and promising concept in regard to hydrolysis properties, lipophilicity of different target structures and application of the concept on various biologically active nucleosides.
Article in the newspaper "Die Welt" on nucleoside diphosphate prodrugs (03.11.2008):
http://www.welt.de/welt_print/article2664791/Trojanische-Pferde-gegen-Aids-und-Hepatitis.html
Selected publications
- C. Meier, CycloSal-Phosphates as Chemical Trojan Horses for the Intracellular Nucleotide and Glycosylmonophosphate Delivery – Chemistry meets Biology, Eur. J. Org. Chem. 2006, 1081-1102.
- C. Meier, A. Meerbach, J. Balzarini, cycloSal-Pronucleotides – Development of First and Second Generation Chemical Trojan Horses for Antiviral Chemotherapy, Frontiers in Bioscience 2004, 9, 873-890.
- C. Meier, M. Ruppel, D. Vukadinovic, J. Balzarini, “Lock-in”-cycloSal-pronucleotides – A New Generation of Chemical Trojan Horses, Minirev. Med. Chem. 2004, 4, 383-394.
- J. Balzarini, S. Aquaro, T. Knispel, C. Rampazzo, V. Bianchi, C.-F. Perno, E. De Clercq, C. Meier, CycloSaligenyl-2‘,3‘-Didehydro-2‘,3‘-dideoxythymidine Monophosphate (cycloSal-d4TMP): Efficient Intracellular Delivery of d4TMP, Mol. Pharmacol. 2000, 58, 928-935.
- C. Meier, A. Lomp, A. Meerbach, P. Wutzler, CycloSal-BVDUMP-Pronucleotides – How to Convert an anti-EBV-inactive Nucleoside Analogue into a Bioactive Compound, J. Med. Chem. 2002, 45, 5157-5172.
- C. Meier, T. Knispel, E. De Clercq, J. Balzarini, CycloSal-Pro-Nucleotides (cycloSal-NMP) of 2',3'-dideoxyadenosine (ddA) and 2',3'-dideoxy-2',3'-didehydroadenosine (d4A): Synthesis and Antiviral Evaluation of a Highly Efficient Nucleotide Delivery System, J. Med. Chem. 1999, 42, 1604-1614.
- C. Meier, T. Knispel, V. E. Marquez, M. A. Siddiqui, E. De Clercq, J. Balzarini, CycloSal-Pro-Nucleotides of 2'-fluoro-ara- and 2'-fluoro-ribo-2',3'-dideoxyadenosine (F-ara- and F-ribo-ddA) as a Strategy to Bypass a Metabolic Blockade, J. Med. Chem. 1999, 42, 1615-1624.
- A. Sauerbrei, C. Meier, A. Meerbach, P. Wutzler, Inhibition Efficiency of cycloSal-Nucleoside Monophophates of Aciclovir and Brivudine on DNA Synthesis of Orthopoxviruses, Antiviral Chem. Chemother. 2006, 17, 25-31.
- C. Meier, U. Görbig, C. Müller, J. Balzarini, CycloSal-PMEA and cycloAmb-PMEA – Potentially new Phosphonate Prodrugs on the Basis of the cycloSal-Pronucleotide Approach, J. Med. Chem. 2005, 48, 8079-8086.
- C. Meier, M. F. H. Ruppel, D. Vukadinović, J. Balzarini, Second Generation of cycloSal-Pronucleotides with Esterase-Cleavable Sites: The „Lock-in“-Concept, Nucleosides, Nucleotides Nucleic Acids 2004, 23, 89-115.
- C. Meier, C. Ducho, H. Jessen, D. Vukadinović-Tenter, J. Balzarini, Second-Generation cycloSal-d4TMP Pronucleotides Bearing Esterase-Cleavable Sites – The „Trapping“ Concept, Eur. J. Org. Chem. 2006, 197-206.
- H. J. Jessen, J. Balzarini, C. Meier, Intracellular Trapping of cycloSal-Pronucleotides: Modification of Prodrugs with Amino Acid Esters, J. Med. Chem. 2008, 51, 6592-6598.
- N. Gisch, J. Balzarini, C. Meier, Studies on Enzyme-cleavable Dialkoxymethyl-cycloSaligenyl-2’,3’-dideoxy-2’,3’-didehydrothymidine Monophosphates, J. Med. Chem. 2008, 51, 6752-6760.
- N. Gisch, F. Pertenbreiter, J. Balzarini, C. Meier, 5-(1-Acetoxyvinyl)-cycloSaligenyl-2’,3’-dideoxy-2’,3’-didehydrothymidine Monophosphates – A Second Type of New, Enzymatically Activated cycloSal-Pronucleotides, J. Med. Chem. 2008, 51, 8115-8123.
- C. Ducho, U. Görbig, S. Jessel, N. Gisch, J. Balzarini, C. Meier, Bis-cycloSal-d4T-monophosphates – Drugs that Deliver Two Molecules of Bioactive Nucleotides, J. Med. Chem. 2007, 50, 1335-1346.
- N. Gisch, J. Balzarini, C. Meier, Doubly loaded cycloSaligenyl-Pronucleotides – 5,5’-Bis-(cycloSaligenyl-2’,3’-dideoxy-2’,3’-didehydrothymidine monophosphates), J. Med. Chem. 2009, 52, 3464-3473.
- H. J. Jessen, T. Schulz, J. Balzarini, C. Meier, Bioreversible protection of nucleosidediphosphates, Angew. Chem. Int. Ed. Engl. 2008, 47, 8719-8722.