Research
Our current research is centred on different aspects of modern catalysis and synthetic organic chemistry. There are two major areas of interest:
Reaction methodology and catalysis
These investigations involve the development of novel and innovative catalytic transformations, particularly, in the area of oxidation catalysis, sequential reactions, and rearrangements. The general aim is to develop novel strategies for catalyst modulation and reactivity tuning. The understanding of factors to control a catalyst is employed to invent new tactics and unusual strategies for the selective functionalization of organic compounds. Starting materials range from very simple organic molecules to somewhat more complex renewables (such as terpenes, fatty acids, and carbohydrates) to densely functionalized natural products.
Target oriented synthetic organic chemistry
These projects are aiming at the construction of functional organic molecules. Target structures in this area are natural products, natural product derivatives and biologically active artificial compounds.
Moreover, novel hybrid natural products and structural and functional conjugates with tailored (bio-)-chemical properties are investigated. The combination of different molecular subunits produces novel molecular architectures with unique characteristics. The exceptional properties can be attributed to synergistic effects of different molecular subunits. Thus, we hope to be able to influence chemical and biochemical properties by modulation of conformation, recognition, transport, and solubility.
Selected Projects
Reaction methodology and catalysis
Oxidative Cyclization
One central area of research in our group is the direct oxidative cyclization of 1,5-dienes and related substrates. This catalytic oxidation allows for the efficient and highly diastereoselective synthesis of oxygen heterocycles. Four new C-O-bonds and up to four stereogenic centers are generated in this single step transformation. For example, the direct oxidative cyclization of 1,5-dienes results in the formation of 2,5-disubstituted tetrahydrofurans (THFs).

• S. Roth, S. Göhler, H. Cheng, C. B. W. Stark, Eur. J. Org. Chem. 2005, 4109-4118. • S. Göhler, S. Roth, H. Cheng, H. Göksel, A. Rupp, L. O. Haustedt,
C. B. W. Stark, Synthesis 2007, 2751. • J. Adrian, S. Roth, C. B. W. Stark, ChemCatChem 2016, 8, 1679-1684.
A specifically optimized variant of this strategy can also be employed for the oxidative mono-cyclization of 1,5,9-trienes and polyenes. The poly-unsaturated substrates undergo mono-cyclization with a high degree of diastereo- and positionselectivity to produce mono-tetrahydrofuran diols with a varying degree of unsaturation. The remarkable positionselectivity appears to be a result of relative electronic properties of the double bonds within the polyolefinic substrates in conjunction with conformational constraints.

• S. Göhler, C. B. W. Stark, Org. Biomol. Chem. 2007, 5, 1605-1614.
1,6-Dienes as starting materials lead to tetrahydropyran (THP) products. Again, good yields and high levels of diastereocontrol are observed. Now 2,6-trans-substituted oxygen heterocycles with one equatorial and one axial substituent are obtained. The C2-symmetric THPs are useful intermediates for the synthesis of natural products such as pheromones.
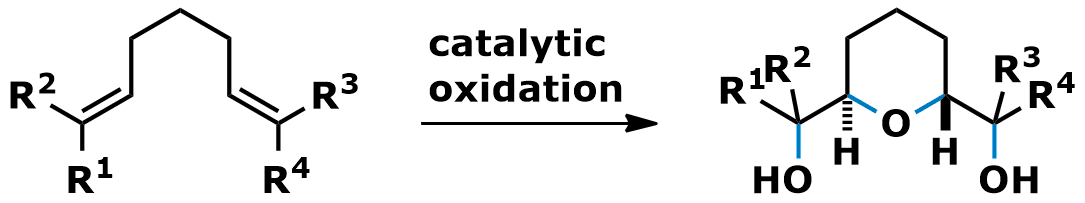
• S. Roth, C. B. W. Stark, Angew. Chem. 2006, 45, 6218-6221.
With a particularly mild catalyst the stereocontrolled oxidative cyclization of 5,6-dihydroxy alkenes to yield THF-diols in high diastereo- and enantiopurity can be achieved. Up to two new chiral centers are created in these cyclization reactions. Many functional groups are tolerated including remote double bonds. The reactions involves a double donor activated ruthenium catalyst. This dual activation is required for the productive intreraction with the proximal double bond.
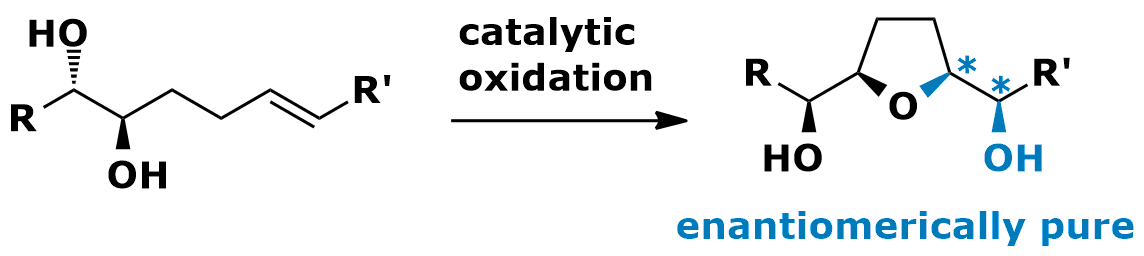
• H. Cheng, C. B. W. Stark, Angew. Chem. 2010, 49, 1587-1590.
Oxidative Degradation
The selective cleavage of carbon–carbon (single) bonds represents an important transformation in organic synthesis. Cleavage reactions in general often provide an unusual and complementary entry to certain functional groups and are therefore of specific interest. For instance, the glycol cleavage and Criegee oxidation are powerful tools for the conversion of vicinal diols to the corresponding aldehydes. The principally related cleavage of 1,2-hydroxy ethers, however, has rarely been studied. Recently, we were able to develop a novel catalytic method for the oxidative cleavage of this motif. Thus, cyclic esters are available starting from cyclic ethers. The lactones are usually obtained in excellent yields.
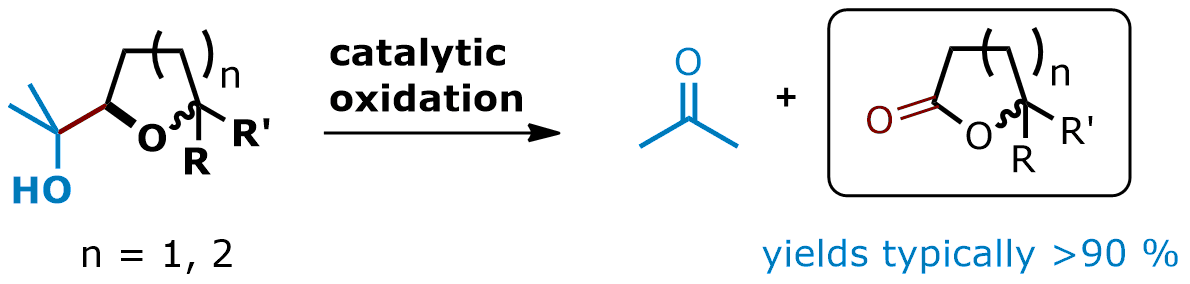
• S. Roth, C. B. W. Stark, Chem. Commun. 2008, 6411-6413.
Alcohol Oxidation
The direct oxidation of primary alcohols to carboxylic acids is an allegedly simple organic reaction. In complex target molecule synthesis, however, no one seems to apply any of the single step procedures. In natural product total synthesis, for instance, the two step sequence (oxidation of the primary alcohol to the aldehyde followed by a second separate oxidation of the aldehyde to the carboxylic acid) seems to be by far the preferred process. In principle, both oxidation steps could proceed through similar intermediates. In our opinion one of the most important causes why the direct oxidation is not effective is the inefficient formation of the intermediary aldehyde hydrates. We therefore initiated a program in search for reagents and condition to stabilize these crucial intermediates and found that N-oxides are quite powerful hydrate stabilizers. On the basis of this discovery we developed a simple, mild and highly effective method for the direct oxidation of primary alcohols to carboxylic acids.
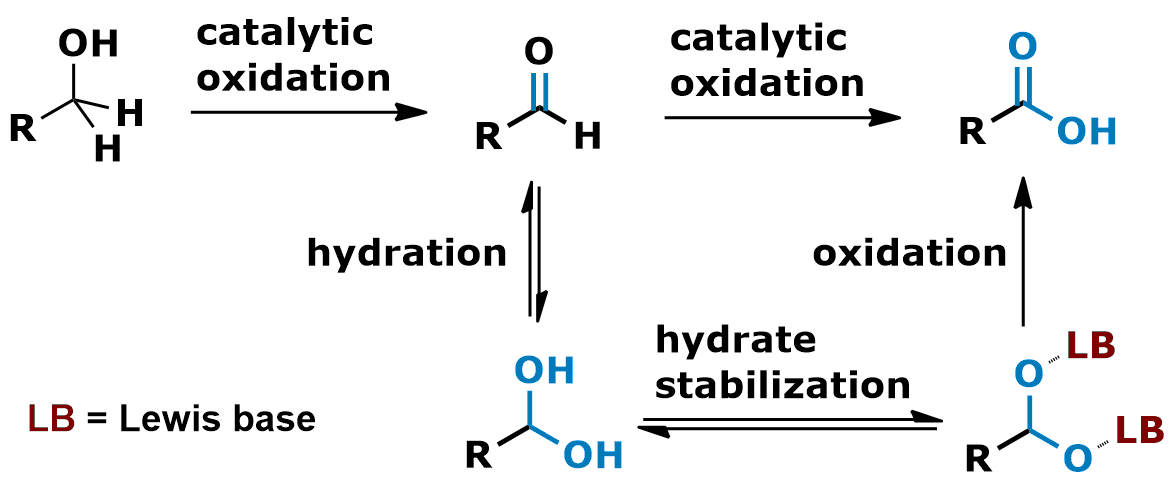
• A.-K. C. Schmidt, C. B. W. Stark, Org. Lett. 2011, 13, 4164–4167. • A.-K. C. Schmidt, C. B. W. Stark, Org. Lett. 2011, 13, 5788–5791. • A. J. K. Roth, M. Tretbar, C. B. W. Stark, Chem. Commun. 2015, 51, 14175–14178.
Natural Product Synthesis
Total Synthesis of cis-Solamin
The annonaceous acetogenins are a class of more than 400 natural products isolated from the tropical plant family Annonaceae. This class of natural products has attracted particular attention due to their remarkable range of biological effects. They exhibit high antitumor, antimalarial, pesticidal, and immunosuppressive activity. Interaction with mitochondrial complex I of the respiratory chain appears to be the molecular basis for at least some of these effects. Using the direct oxidative cyclization as a crucial transformation an enantioselective total synthesis of cis-solamin, a typical member of this class of natural products, has been accomplished. Further key steps involve an enzymatic desymmetrization, a TPAP-catalyzed oxidative termini differentiation, and a ruthenium-catalyzed Alder-ene reaction. Thus, the total synthesis of cis-solamin was achieved in 11 steps with an overall yield of 7.5%.
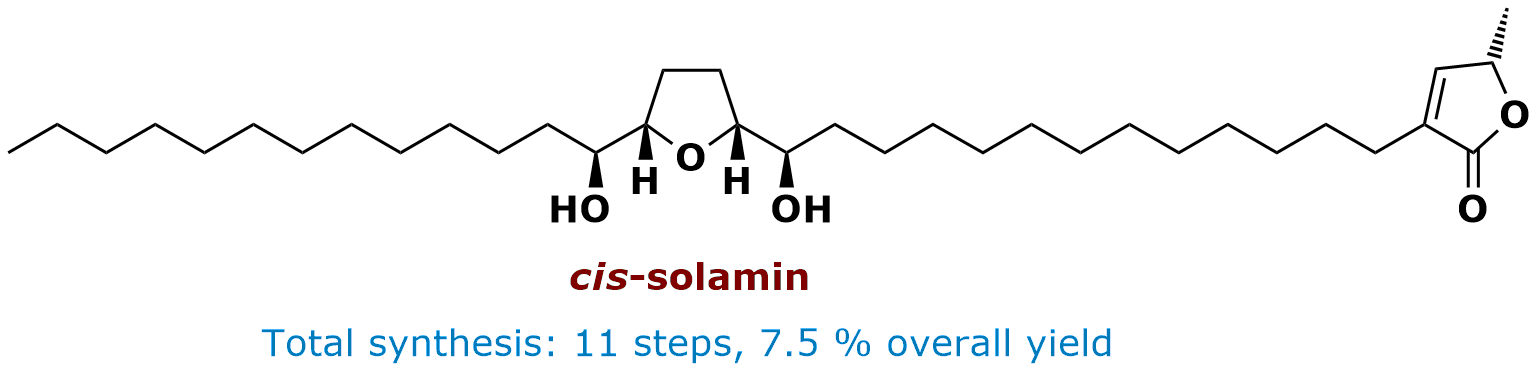
• H. Göksel, C. B. W. Stark, Org. Lett. 2006, 8, 3433-3436. • Highlighted in Synfacts 2007, 0012.
Total Synthesis of Muricadienin
A certain subgroup of annonaceous acetogenins does not contain the otherwise characteristic THF (or THP) rings but C-C-double bonds instead. In fact, these (poly)unsaturated compounds are believed to be the biosynthetic precursors of the THF containing acetogenins. The putative precursor of cis-solamin is a natural product named muricadienin. We were able to achieve the first total synthesis in 11 steps with a remarkable overall yield of 42%.

• J. Adrian, C. B. W. Stark, Org. Lett. 2014, 16, 5886-5889.
Biomimetic Total Synthesis of Heronapyrrole C
In late 2010, Capon and co-workers from the University of Queensland (Australia) reported the isolation and structural elucidation of a set of unique nitropyrroloterpenes named heronapyrrole A-C. We quickly became interested in these bioactive bacterial metabolites (Streptomyces sp. CMB-M0423) and decided to help investigating the stereostructure – which was unassigned at that time – of heronapyrrole C through total synthesis. Our biomimetic approach and proposal of relative and absolute stereochemistry of heronapyrrole C was reported in July 2012.
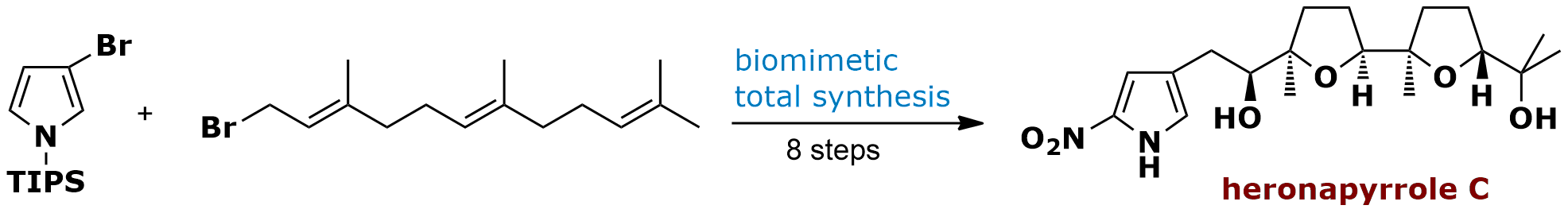
• J. Schmidt, C. B. W. Stark, Org. Lett. 2012, 14, 4042-4045. • J. Schmidt, C. B. W. Stark, J. Org. Chem. 2014, 79, 1920-1928.